原子力基盤技術データベースのメインページへ
作成: 2000/02/25 蕪木 英雄
データ番号 :190025
分子動力学法による固体の熱伝導シミュレーション
目的 :固体の熱伝導率の数値シミュレーションによる予測
研究実施機関名 :日本原子力研究所 数値実験技術開発グループ
応用分野 :材料、統計物理、計算物理
概要 :
工学的な目的のためにある材料を用いて熱、構造計算のような現象論的式に基づき計算を行う時、固体の物性値は入力パラメータとして重要な働きをする。通常物性値は実験で測定されるが、物性値の温度依存性、極端な条件下での詳細なデータを求めることは困難を伴うことが多い。そのため、ミクロな粒子法を用いた数値シミュレーションにより出来るだけ仮定を用いないで固体の熱伝導率を予測する手法を確立した。
詳細説明 :
液体の輸送係数を計算するために古典的な分子動力学を用いることは、信頼できる原子間のポテンシャルが与えられる場合は既に確立した技術である。特に、希ガス系のシステムにたいしてはレナード・ジョーンズポテンシャルは長い間基準モデルとして使用されてきた。しかし、定量的な解析を行なう目的には、多体及び量子効果を無視することが出来ないことは良く知られてきた。液体の熱伝導に関しては、レナード・ジョーンズポテンシャルは実験と良い一致を与えることが知られている。それゆえ、固体アルゴンについてもこのレナード・ジョーンズポテンシャルを用いることで良い結果が得られることが期待される。
液体状態に対して、固体の熱伝導を分子動力学を用いて解析した例は非常に少ない。現在まで、これらのどの仕事も分子動力学による手法が実験データに対して定量的に結果を与えるかを明確に示していない。一方、希ガス結晶の熱伝導率の解析においては、フォノン散乱によるいくつかの試みが報告されているが、実験結果との一致については結論は出ていない。これは理論計算に近似が含まれているので、理論的仮定とポテンシャルモデルの妥当性を区別することが出来ないことに原因がある。
数値シミュレーションでは平衡系の分子動力学法を用い、熱伝導率はGreen-Kuboの方法に基づき熱流束の相関関数< J(t)J(0)>を積分することにより導出する。ここでは、アルゴン単結晶の熱伝導率を計算した結果について報告する(原論文1)。6-12レナード・ジョーンズポテンシャル(ε=119.8[K]、 σ=3.405[Å])により相互作用するN個(N=256)のアルゴン原子を含む周期境界を持つ領域を考え、ニュートンの運動方程式を5次精度の予測子-修正子(Gear)法で時間積分した。
一定圧力(P=0)下において、温度10Kから融点近傍までの熱伝導率を数値シミュレーションにより求めた結果を図1に示す。この結果をT-nにフィットすると、n=1.5となり、実験結果のフィットの値と良く一致することが分かった。熱伝導率の絶対値については、実験結果は測定全温度範囲で一様にレナード・ジョーンズによる計算結果より1.9倍大きいことが分かった。この不一致は、大部分原子間ポテンシャルモデルが悪いことに起因している。液体のシミュレーションで良い結果を得ているレナード・ジョーンズポテンシャルは実験値より低い値を得ること、即ち非調和性が強過ぎることが明らかになった。一方、改良された第一原理に基づくポテンシャルを用いることにより熱伝導の絶対値は良く予測出来ることが明らかになった(参考資料1)。
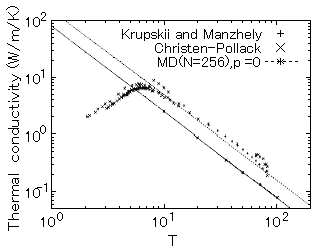
図1 固体アルゴンの熱伝導率の温度依存性(原論文1より引用)
この手法では、熱伝導率の値の他に、相関関数の時間による減衰のデータから熱伝導の動的な性質について調べることが可能である。われわれの結果では、融点近傍では熱流束に対する相関関数は単一の指数関数的過程で減衰することが分かった。また、温度が下がりデバイ温度の半分以下になると、第二の(よりゆっくりした)緩和成分が出現することを発見した(図2)。図2により温度が低くなるにつれ第2の緩和過程が出現してくることがより明瞭にみることができる。これら初期の速い減衰は局所的なダイナミクス、遅い減衰は格子振動によるフォノン輸送のダイナミクスに起因するものと考えられる。
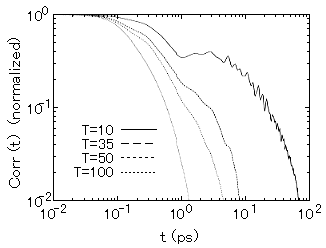
図2 時間相関関数の緩和過程における第二の成分の温度依存性(原論文1より引用)
現在行なわれている熱伝導の理論解析が熱伝導率を直接計算するかまたは有効な緩和時間を計算しており、ダイナミクスの情報は温度依存性を通して間接的にしか表れないのに比較して、ここで行ったGreen-Kuboの方法に基づく平衡分子動力学手法では、緩和のダイナミクスをそのまま取り扱うことができ、その中でフォノンの果たす役割を明確に出来るものと考えている。
コメント :
原論文1 Data source 1:
Thermal conductivity of solid argon by classical molecular dynamics
H. Kaburaki, J. Li(*), S. Yip(*)
Japan Atomic Energy Research Institute; (*)Massachusetts Institute of Technology
Mat. Res. Soc. Symp. Proc. Vol. 538, (1999) pp. 503-508.
参考資料1 Reference 1:
H. Kaburaki, J. Li(*), S. Yip(*)
Japan Atomic Energy Research Institute,(*)Massachusetts Institute of Technology
to be published.
キーワード:分子動力学法、熱伝導、固体、レナード・ジョーンズポテンシャル、アルゴン、グリーン・久保の方法、相関関数、予測子ー修正子法
molecular dynamics method, thermal conduction, solid, Lennard-Jones potential, argon, Green-Kubo method, correlation function, predictor-corrector method
分類コード:190204
原子力基盤技術データベースのメインページへ